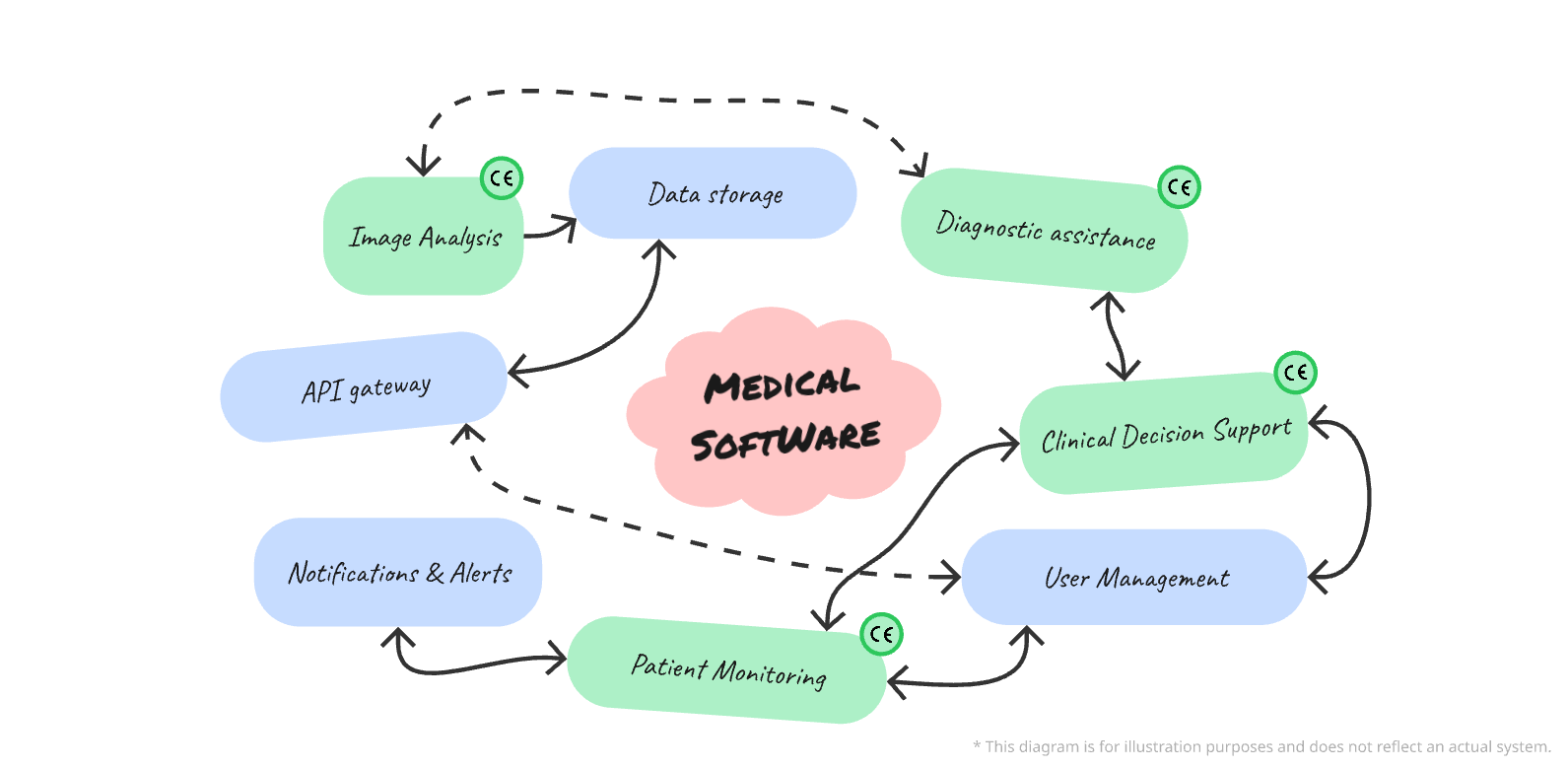
Gestion des fonctionnalités d’un dispositif médical numérique
L’obtention du marquage CE pour un logiciel en tant que dispositif médical (DM) est une étape cruciale pour sa mise sur le marché européen. Cependant, tous les logiciels n’ont pas de fonctionnalités à vocation médicale. La distinction entre fonctionnalités DM et non-DM (non dispositif médical) est essentielle pour garantir la conformité aux exigences du règlement (UE) 2017/745 (MDR) et pour assurer une documentation réglementaire précise. Dans cet article, nous vous expliquons comment gérer cette séparation et documenter correctement ces fonctionnalités dans le cadre de l’obtention du marquage CE.
Comprendre les fonctionnalités DM vs non-DM
La première étape consiste à comprendre ce qui constitue une fonctionnalité dispositif médical.
Selon l’article 2 du MDR 2017/745, un dispositif médical est défini comme « tout instrument, appareil, équipement, logiciel, implant, réactif in vitro ou tout autre article destiné par le fabricant à être utilisé, seul ou en combinaison, à des fins médicales, telles que le diagnostic, la prévention, le suivi, le traitement ou l’atténuation d’une maladie, d’une blessure ou d’un handicap ».
Ainsi, un logiciel n’est qualifié de dispositif médical que si ses fonctionnalités ont une finalité médicale clairement définie. À l’inverse, les fonctionnalités non-DM sont celles qui n’ont pas d’objectif médical, telles que les outils administratifs, de gestion ou de communication qui ne sont pas utilisés pour influencer une décision médicale.
Exemples de Fonctionnalités DM :
- Logiciels d’aide au diagnostic à partir d’imagerie médicale.
- Applications de surveillance des paramètres vitaux.
- Outils d’aide à la décision clinique.
Exemples de Fonctionnalités Non-DM :
- Gestion de la facturation
- Systèmes de planification ou de gestion des ressources humaines
Séparation des fonctionnalités DM et Non-DM
Il est crucial de bien séparer les fonctionnalités DM et non-DM pour plusieurs raisons :
- Conformité réglementaire : Seules les fonctionnalités DM doivent respecter les exigences du MDR, y compris la gestion des risques, la validation clinique et le marquage CE. Les fonctionnalités non-DM ne sont pas soumises aux mêmes obligations.
- Gestion des risques : Une séparation claire permet de gérer les risques de manière ciblée, en se concentrant sur ceux qui concernent les fonctionnalités DM.
- Clarté dans la documentation : La documentation technique et le dossier de marquage CE doivent clairement indiquer quelles parties du logiciel sont soumises à la réglementation.
Le guide MDCG 2019-11 précise que la qualification d’un logiciel en tant que dispositif médical doit être fondée sur l’intention médicale de chaque fonction. Ce guide fournit des critères et des exemples pour déterminer si un logiciel relève de la réglementation des dispositifs médicaux ou non.
Il indique notamment que, « un logiciel qui vise à améliorer ou gérer des conditions médicales, comme la détection, l’analyse, la prévision de maladies, ou la gestion des soins d’un patient, est un dispositif médical** »**. En revanche, un logiciel utilisé pour des fins administratives ou de gestion sans finalité médicale ne sera pas considéré comme un DM.
Comment documenter cette séparation dans le dossier technique ?
La documentation est un élément clé pour garantir la conformité de votre logiciel. Voici comment structurer et documenter cette séparation :
1. Description du produit
Dans le dossier technique, la description du produit doit clairement distinguer les modules ayant une finalité médicale de ceux qui n’en ont pas. Chaque module doit être documenté avec son objectif et ses exigences spécifiques.
2. Analyse des exigences essentielles (GSPR)
Les exigences des annexes I du MDR s’appliquent uniquement aux fonctionnalités DM. Dans votre dossier technique, il convient de lister et d’expliquer quelles exigences s’appliquent à chaque fonctionnalité. Par exemple, la gestion des risques, la validation clinique et la traçabilité des données seront requises pour les fonctionnalités à finalité médicale. Pour les modules non-DM, ces exigences ne seront pas pertinentes et seront indiquées comme « hors champ ».
3. Analyse des risques
L’analyse des risques dans le cadre des logiciels de dispositifs médicaux, même pour les fonctionnalités non-DM, constitue une bonne pratique afin de garantir que le fonctionnement global du système n’affecte pas la sécurité ni la performance des fonctionnalités DM.
Il est essentiel d’évaluer l’impact potentiel des fonctionnalités non-DM sur les modules DM. En particulier, il convient de vérifier que ces fonctionnalités non-DM n’interfèrent pas avec les fonctionnalités DM. Par exemple, si un dysfonctionnement dans un module non-DM compromettait la sécurité ou la performance d’un module DM, cet impact doit être pris en compte dans l’analyse des risques.
Justification de l’exclusion des fonctionnalités non-DM du marquage CE
Lorsqu’un logiciel combine des fonctionnalités DM et non-DM, il est essentiel de justifier clairement pourquoi certaines parties du logiciel ne sont pas soumises au marquage CE. Cette justification repose sur l’absence de finalité médicale des fonctionnalités non-DM. Par exemple, un module d’archivage de données peut être considéré comme un outil général sans finalité médicale, et donc non soumis au marquage CE. Cette distinction doit être faite de manière explicite dans le dossier technique.
Pour faciliter cette séparation, il est recommandé de créer des modules logiciels indépendants pour les fonctionnalités DM et non-DM.
Le guide MDCG 2019-11 souligne également l’importance de l’architecture modulaire pour garantir la conformité du logiciel. Il conseille de maintenir une séparation nette entre les modules fonctionnels, en particulier lorsque certaines parties du logiciel ne sont pas soumises au marquage CE, afin de limiter les risques et de simplifier le processus d’audit.