Medical device feature management
Obtaining the CE marking for software as a medical device (MD) is a crucial step for market access in Europe. However, not all software features are intended for medical purposes. Distinguishing between MD and non-MD features is essential to ensure compliance with the requirements of Regulation (EU) 2017/745 (MDR) and to maintain accurate regulatory documentation. This article explains how to manage this distinction and properly document these features as part of the CE marking process.
Understanding MD vs non-MD features
The first step is understanding what constitutes a medical device feature.
According to Article 2 of MDR 2017/745, a medical device is defined as “any instrument, apparatus, appliance, software, implant, reagent for in vitro use, or any other article intended by the manufacturer to be used, alone or in combination, for medical purposes such as diagnosis, prevention, monitoring, treatment, or alleviation of disease, injury, or disability.”
Thus, a software is classified as a medical device only if its features have a clearly defined medical purpose. Conversely, non-MD features are those that lack a medical objective, such as administrative, management, or communication tools that are not used to influence medical decisions.
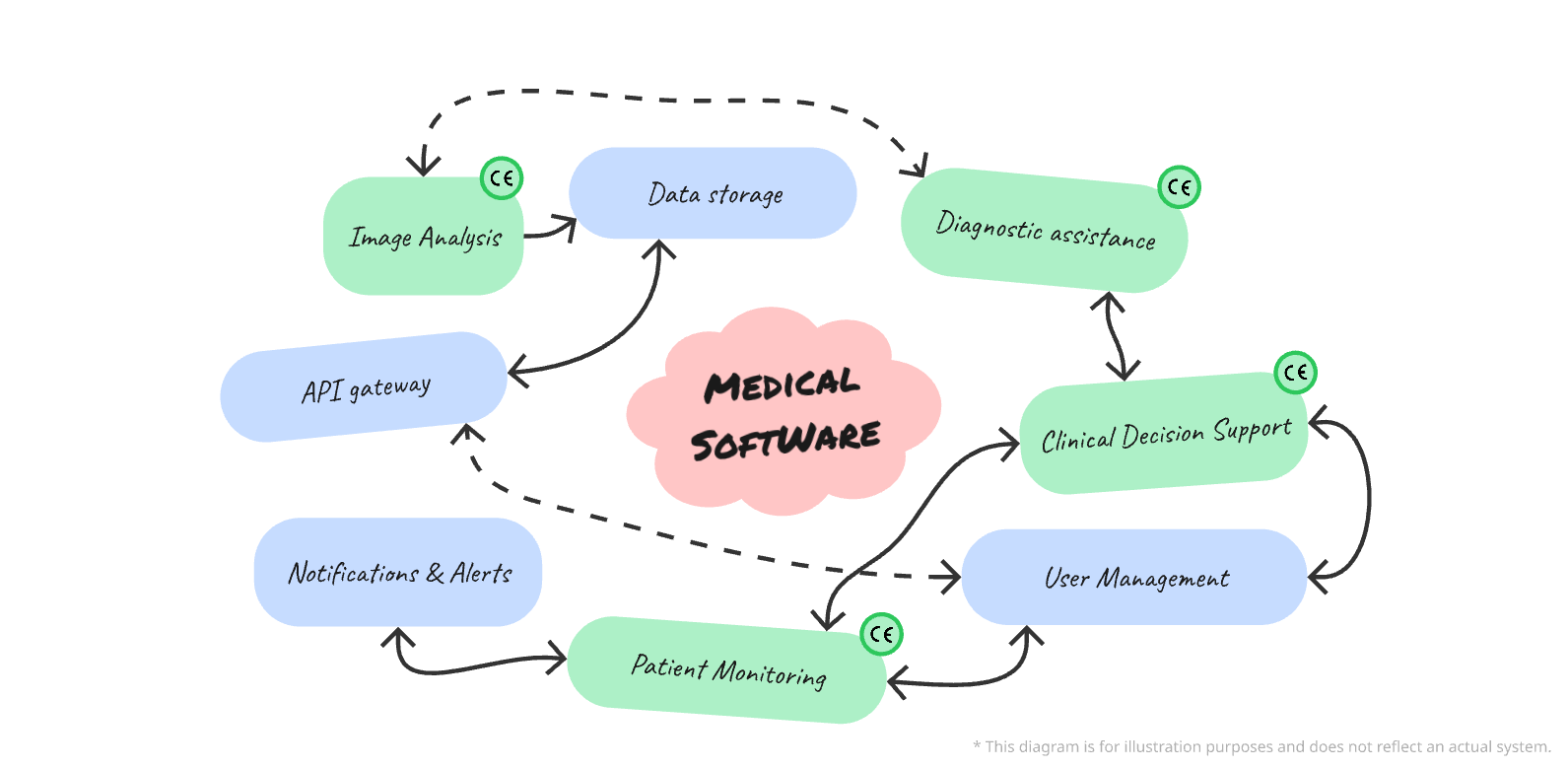
Examples of MD Features:
- Diagnostic support software from medical imaging.
- Vital signs monitoring applications.
- Clinical decision support tools.
Examples of Non-MD Features:
- Billing management.
- Scheduling or human resources management systems.
Separation of MD and non-MD features
It is critical to clearly separate MD and non-MD features for several reasons:
- Regulatory Compliance: Only MD features must comply with MDR requirements, including risk management, clinical validation, and CE marking. Non-MD features are not subject to these obligations.
- Risk Management: Clear separation allows targeted risk management, focusing on risks related to MD features.
- Clarity in Documentation: The technical documentation and CE marking dossier must clearly indicate which parts of the software are subject to regulation.
The MDCG 2019-11 guidance specifies that software qualification as a medical device should be based on the medical intent of each function. The guidance provides criteria and examples for determining whether software is subject to medical device regulations.
It states, for example, that “software aimed at improving or managing medical conditions, such as disease detection, analysis, prediction, or patient care management, is a medical device.” Conversely, software used for administrative or management purposes without a medical aim is not considered an MD.
How to document this separation in the technical file
Documentation is key to ensuring your software’s compliance. Here’s how to structure and document this separation:
1. Product Description
In the technical file, the product description must clearly distinguish modules with medical purposes from those without. Each module should be documented with its objective and specific requirements.
2. Essential Requirements Analysis (GSPR)
The requirements of Annex I of the MDR apply only to MD features. In your technical file, list and explain which requirements apply to each feature. For example, risk management, clinical validation, and data traceability will be required for MD features. For non-MD modules, these requirements are not applicable and should be marked as “out of scope.”
3. Risk analysis
Conducting a risk analysis for both MD and non-MD features is good practice to ensure the overall system’s operation doesn’t affect the safety or performance of MD features. It’s crucial to evaluate the potential impact of non-MD features on MD modules. Specifically, check that non-MD features do not interfere with MD functionality. For example, if a malfunction in a non-MD module could compromise the safety or performance of an MD module, this impact must be considered in the risk analysis.
Justification for excluding non-MD features from CE marking
When software combines MD and non-MD features, it is essential to clearly justify why certain parts of the software are not subject to CE marking. This justification is based on the lack of medical purpose in non-MD features. For instance, a data archiving module may be considered a general tool without medical intent and thus not subject to CE marking. This distinction should be explicitly made in the technical file.
To facilitate this separation, it is recommended to create independent software modules for MD and non-MD features.
The MDCG 2019-11 guide also emphasizes the importance of a modular architecture to ensure software compliance. It advises maintaining a clear separation between functional modules, especially when some parts of the software are not subject to CE marking, to reduce risks and simplify the audit process.